- Reasons for development
- Features of neuroendocrine tumors
- Classification of neuroendocrine tumors
- Types of neuroendocrine tumors and their symptoms
- Diagnosis of the disease
- Treatment
- Survival prognosis
Neuroendocrine tumors (NETs) are formed from apudocytes , or APUD cells . These cells are scattered throughout the body and constitute the most ancient part of the endocrine system. They are at the same time similar to nerve cells and cells of the endocrine glands, since they can respond to signals from the outside or changes in the state of the body, and are capable of producing hormones that perform different functions.
Tumors of the APUD system are rare and can be difficult to diagnose. They most often occur in the gastrointestinal tract, but can also affect other organs. The peculiarity of neuroendocrine neoplasms is that tumor cells produce an increased amount of hormones, and because of this, certain symptoms may occur.
According to the American SEER registry, in 2004 the incidence of neuroendocrine tumors in the United States was 5 cases per 100 thousand population. In Russia, unfortunately, there are no statistics, but the incidence is probably at a similar level.
According to American experts, the prevalence of neuroendocrine tumors is growing every year. This is primarily associated with changes in nutritional patterns and unfavorable environmental conditions.
According to the same SEER registry, neoplasms of the APUD system are often diagnosed at late stages: in 50% of cases, the tumor manages to spread to surrounding tissues, to regional lymph nodes, and give distant metastases.
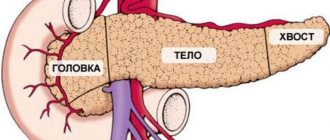
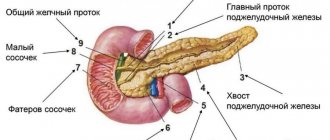
What is a neuroendocrine tumor of the pancreas?
Pancreatic neuroendocrine neoplasms (PNEN) are a group of neoplasias (tumors) of different origin, structure and clinical manifestations.
Depending on whether these tumors can secrete hormones and amines, causing carcinoid and other clinical syndromes, they can be classified as functional (F-NEN) or nonfunctional (NF-NEN). Neuroendocrine neoplasms of the pancreas are the most common neuroendocrine tumors.
Most PNENs are low grade but have malignant potential. They grow slowly and remain stable for many years, at least when they are small. However, without treatment, most of these tumors grow and eventually metastasize to the liver.
Pancreatic neoplasms are rare overall: <2% of all pancreatic tumors. Their diagnosis and localization often require significant technology, and their successful treatment requires a thorough multidisciplinary approach.
For these reasons, a patient with a possible or confirmed pancreatic endocrine tumor is best treated in a large multidisciplinary center that includes endocrinologists, gastroenterologists, pancreatic surgeons, and medical oncologists.
Features of neuroendocrine tumors
Neuroendocrine cells are not associated with any specific organ. They are scattered throughout the body. Although tumors most often occur in the digestive tract, in more rare cases they are found in other places. In 15% of cases, the primary tumor cannot be found.
Neuroendocrine tumors can produce hormones that cause certain effects in organs, causing the patient to experience certain symptoms. Such neoplasms are called functioning. If hormones are not produced, it is a non-functioning tumor.
Classification
The term neuroendocrine tumor of the pancreas usually refers to insulinoma, gastrinoma or glucagonoma. Sometimes more rare VIPomas and somatostaniomas are added to this trio.
PNENs are defined as functional or nonfunctional depending on whether they cause a syndrome of hormonal hypersecretion—excessive production of hormones.
Of all functional PNENs, gastrinomas and insulinomas are the most common. Other neoplasias are usually considered together as a group called "rare functional PNENs."
Several such rare clinical syndromes have been proposed as possible functional PNENs. These include the following:
- calcitoninoma;
- parathyrinoma;
- growth hormone-secreting tumor (GRFoma);
- adrenocorticotropin hormone-secreting neoplasia (ACTHoma);
- neurotensinoma.
According to European and American oncologists, non-functional PNEN are more common than functional ones. The former are usually diagnosed in the fourth or fifth decades of life and often have already metastasized to the liver at the time of diagnosis.
Reasons for appearance
10% of functional NENs become a manifestation of hereditary familial endocrine tumors:
- multiple endocrine neoplasia syndrome type 1 (MEN1);
- von Hippel-Lindau disease (VHL);
- neurofibromatosis type 1 (NF-1);
- tuberous sclerosis (TSC).
The reason for the development of these pancreatic neuroendocrine neoplasms is hereditary switching off of the corresponding tumor suppressor gene during fetal development.
Although the outdated term “islet cell tumor” is often used today to identify neoplasms of the endocrine pancreas, it is a misnomer. Many pancreatic neuroendocrine tumors do not develop directly from islet cells.
Most of these neoplasms arise from APUD stem cells. These are the precursor cells from which all types of pancreatic tissue are subsequently formed. They are located in the epithelium of the ducts of this organ.
The fact that many gastrinomas and somatostatinomas are located near the glandular tissue of the pancreas, but not within it, supports the view that these neoplasms may develop extrapancreatically.
Types of neuroendocrine tumors and their symptoms
Since there are different types of neuroendocrine tumors and they can be located in different organs, the symptoms vary. Three main groups of manifestations can be distinguished. The first is the general symptoms characteristic of any type of cancer: weakness, increased fatigue, decreased appetite, weight loss for no apparent reason.
The second group of symptoms is associated with the location of the tumor and its size, compression of anatomical structures and dysfunction of the affected organ. Concerns include pain in a certain part of the body, nausea, persistent chronic cough, stool and urination disorders, bleeding, and unusual discharge. When the liver, bile ducts, and pancreas are damaged, obstructive jaundice develops.
The third group of symptoms is caused by hormones produced by neuroendocrine cells:
- diarrhea - loose stools more than three times a day;
- constant thirst, hunger, frequent urination are signs of increased blood glucose levels;
- increased fatigue, irritability, trembling, dizziness, convulsions, loss of consciousness are signs of a decrease in blood glucose levels;
- peptic ulcer that cannot be treated;
- anxiety;
- skin rash.
Neuroendocrine tumors of the gastrointestinal tract
The gastrointestinal tract is the most common location of neuroendocrine tumors. Such neoplasms are often called carcinoid tumors . Frequency of damage to different parts of the digestive tract:
- small intestine - 39%;
- rectum - 15%;
- vermiform appendix (appendix) - 7%;
- colon - 5–7%;
- stomach - 2–4%.
Tumors in the digestive tract cause classic carcinoid syndrome . It manifests itself in the form of redness and a feeling of warmth in the face, diarrhea, shortness of breath, symptoms reminiscent of bronchial asthma, weakness, rapid heartbeat, weight gain for no apparent reason, high blood pressure, and its frequent fluctuations.
Other neuroendocrine tumors
The second most common site of localization of tumors of the APUD system after NETs of the gastrointestinal tract is the lungs. In approximately 30% of cases, a neuroendocrine neoplasm occurs in the bronchial system.
In 7% of cases, NETs affect the pancreas. In this situation, the term “islet cell tumors” is used.
Pheochromocytoma is a tumor of the adrenal gland (sometimes in other parts of the body) that produces stress hormones: adrenaline and norepinephrine. As a result of excess levels of hormones, attacks (crises) occur, during which a person experiences anxiety, fear, chills, trembling, headache and chest pain, his skin becomes pale, the heartbeat quickens, extrasystoles occur, nausea, and vomiting.
Merkel cell carcinoma is a malignant skin tumor. It is very rare, but is highly aggressive, spreads early to the lymph nodes, and metastasizes. The pathology manifests itself in the form of one or several nodules on the skin of a red and bluish color. They may ulcerate. Merkel cell cancer is difficult to diagnose and is often detected in late stages.
Symptoms of the disease
Functional tumors lead to hormonal hypersecretion syndromes.
| Neoplasia | Symptom(s) |
| Insulinoma | Hypoglycemia is a decrease in blood glucose levels. Sweating, weakness, tremors, hunger, etc. |
| Gastrinoma (ulcerogenic pancreatic adenoma) | It manifests itself as a protracted gastric ulcer that is difficult to treat - abdominal pain, nausea, vomiting, etc. |
| VIPoma | Uncontrollable diarrhea, decreased potassium levels in the blood, decreased acidity of gastric juice, dehydration, weight loss. |
| Glucagonoma | Glucose intolerance, dermatitis, stomatitis, glossitis, deep vein thrombosis, depression, heart failure. |
| Somatostatinoma | Symptoms of diabetes mellitus and cholelithiasis, steatorrhea, achlorhydria. |
Nonfunctional neoplasms cause nonspecific symptoms, such as vague abdominal pain, and are usually discovered incidentally.
The distinction between functional and nonfunctional PNEN is based on clinical presentation, and there is no absolute difference in hormone expression between functional and nonfunctional PNEN.
For example, tumor cells in a small PNEN may express glucagon, resulting in borderline elevated glucagon levels. Clinically, however, this PNEN is defined as nonfunctional because a small increase in glucagon is not sufficient to cause glucagonoma syndrome.
Classification of neuroendocrine tumors
There are different classifications of neuroendocrine tumors. The table below shows the main groups:
| Depending on whether the tumor produces hormones |
|
| Depending on location |
|
| Depending on the degree of differentiation - how much the tumor tissue differs from normal tissue. |
|
| Pancreatic tumors that produce certain hormones |
|
The stage of neuroendocrine tumors is determined in accordance with the generally accepted TNM system:
- T is the size of the primary tumor, its growth into surrounding tissues.
- N - damage to regional (close to the tumor) lymph nodes.
- M—presence of distant metastases.
Classification by stage differs depending on the organ in which the neuroendocrine neoplasm is located. For example, the stage of an APUD tumor in the lung is determined in the same way as for non-small cell lung cancer. For neuroendocrine neoplasms in the stomach, small intestine, appendix, colon, rectum and pancreas, their own stage classification systems have been developed.
Diagnostics
Because recognition of hormonal hypersecretion syndrome requires considerable clinical experience and the symptoms of nonfunctional tumors are nonspecific, the diagnosis of PNEN often occurs quite late.
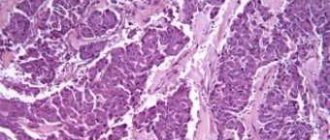
Endocrine testing, imaging, and histologic findings are essential for accurate diagnosis of PNEN.
A complete diagnosis must establish the nature of PNEN, assess tumor grade, identify primary and metastatic sites, and determine whether the tumor is functioning. If hormonal hypersecretion syndrome is suspected, appropriate biochemical testing is performed to determine it, followed by imaging, endoscopy and biopsy.
If a tumor in the pancreas or liver is incidentally identified by imaging, it is usually biopsied to confirm the presence of PNEN. Biochemical testing is ideally performed even if hormonal hypersecretion syndrome is not obvious, as it may be at a subclinical stage, and hypersecreted hormones can be used as tumor markers during subsequent evaluations.
Survival prognosis
The prognosis depends on what type of tumor was diagnosed, how aggressive it is, and what stage it is at. Treatment has one of two goals:
- Completely remove tumor foci. If the examination results do not reveal signs of the presence of a tumor in the body, remission is stated.
- Relieve the patient of symptoms and contain the growth of the tumor. In this case, the patient can live quite a long time.
It is worth talking to your doctor before starting treatment, asking what the goal will be, what results can be expected, and what is planned to be done if the situation worsens and the tumor recurs.
Book a consultation 24 hours a day
+7+7+78
Treatment
Patients with pancreatic neuroendocrine neoplasms are treated individually to balance the effects of hormonal hypersecretion. Many pancreatic endocrine tumor syndromes are potentially life-threatening. Therefore, initial medical therapy is aimed at stabilizing the patient's condition to allow a complete preoperative assessment.
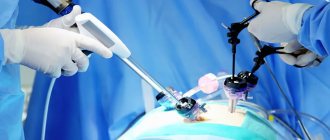
All resectable functional or nonfunctional PNEN are recommended to be treated surgically. Different types of surgical intervention are used:
- tumor enucleation (removal of the visible part of the tumor while preserving healthy organ tissue);
- enucleation in combination with pancreatic resection;
- distal pancreatectomy;
- pancreatoduodenectomy (removal of the pancreas and part of the intestine);
- etc.
The type of surgical intervention is determined individually, based on the characteristics of a particular disease in a particular patient. At the same time, neuroendocrine pancreatic cancer requires mandatory excision of lymph nodes.
All resectable insulinomas should be removed surgically, regardless of tumor size. 85–90% of patients with this type of PNEN can be completely cured in this way. Insulinomas account for approximately 35–40% of functional PNENs. Of these, malignant insulinomas account for only about 5–10% of the total.
Systemic therapy is necessary for patients with residual disease after surgery and for the treatment of inoperable tumors.
Synthetic analogues of the hormone somatostatin (growth hormone) are effective against functional PNEN. Their effect is especially pronounced in VIPomas and glucagonomas.
There is no clear evidence that somatostatin analogues treat nonfunctional PNEN, but they are likely to inhibit tumor growth.
Two somatostatin analogues, octreotide and lanreotide, and their long-acting formulations are currently available, and a new analogue, pasireotide, is still in the clinical trial phase with encouraging preliminary results.
Progressive treatments for neuroendocrine pancreatic neoplasia in Belgium
In clinics in Belgium, modern methods of minimally invasive surgery, radiation therapy, biological therapy and chemotherapy are used in the treatment of PNEN.
Microinvasive surgical and radiological methods include:
- radioactive polymer microspheres;
- chemoembolization;
- mild embolization;
- radiofrequency ablation (RFA);
- percutaneous alcohol ablation;
- microwave ablation.
Systemic interferon-α2A is used in clinics in Belgium for the treatment of PNEN, which are insensitive to somatostatin analogues.
The mTOR inhibitor everolimus (RAD001) and the tyrosine kinase inhibitor sunitinib are effectively used in Belgian clinics to slow the progression of neoplasia.
Chemotherapy is available for intermediate- and high-grade neoplasias. Temozolomide and capecitabine are the drugs of choice that can be used in patients with rapidly progressing PNEN.
The method of Peptide Receptor Radionuclide Therapy (PRRT) Lutetium Lu 177-dota-tate, developed by Belgian scientists in collaboration with colleagues from the Netherlands, is being successfully introduced into practice. It is effective against well-differentiated PNEN. But this method is not yet widely used, so it is best suited for neoplasias with high malignant potential that are resistant to other systemic treatments
Complications
Patients with pancreatic endocrine neoplasia may experience morbidity associated with tumor growth.
- Patients with tumors in the head of the pancreas sometimes have a bile duct blockage, a pancreatic blockage, or both.
- Chronic abdominal pain may occur due to the compressive effect of a large intra-abdominal tumor or obstructive pancreatitis.
Morbidity caused by the effects of excess hormonal production by functional pancreatic neoplasms occurs earlier than the neoplastic effects in the course of the disease.
- If left untreated, patients with insulinoma may experience hypoglycemic seizures and even open coma.
- Before the development of effective antisecretory drugs (eg, H2 blockers and proton pump inhibitors), patients with gastrinoma often experienced life-threatening gastrointestinal bleeding from peptic ulcer disease.
- Patients with VIPomas are at risk of becoming dehydrated from diarrhea. They may also develop fatal cardiac arrhythmias due to associated hypokalemia.
- The morbidity associated with the hormonal effects of glucagonoma can be polymorphic. Patients may have diabetes, malnutrition, stomatitis, and other symptoms similar to severe nutritional deficiencies.
Forecast
The prognosis for most types of PNEN is favorable. But even that neuroendocrine tumor of the pancreas, for which the prognosis is considered good, requires constant monitoring. Because increased growth rate can become an alarming factor that worsens survival.
Because these tumors typically grow slowly and have a relatively low metastatic potential, and because no specific criteria have been defined to predict their behavior, the distinction between benign and malignant neoplasms is based on the presence of metastatic disease.
The five-year survival rate for PNEN without treatment is:
- 80% for all stages;
- 90–100% for localized disease;
- 40% for regional disease;
- 29% for distant metastases.
Long-term disease-specific survival (DSS) of surgically resected tumors exceeds 50% at 20 years.
Because even metastatic pancreatic endocrine neoplasms typically grow slowly, the prognosis of patients with these tumors is relatively good compared with that of patients with nonendocrine cancers.
More than 90% of patients with insulinomas have benign tumors without evidence of metastases, and up to 97% of these patients are completely cured by surgical resection.
Patients with gastrinomas have a worse prognosis. 60% of these tumors are malignant. However, survival rates differ strikingly between patients whose gastrinoma metastases are limited to lymph nodes and those with liver metastases, as follows:
- patients whose gastrinoma metastases involve only lymph nodes may live 25 years or longer, and their life expectancy is indistinguishable from that of people without gastrinoma.
- patients with liver metastases have a 5-year survival rate of 20-30%, compared to 90% for patients without liver metastases.
Approximately half of all VIPomas have metastases at the time of diagnosis or surgery, and approximately one third of patients are completely cured by surgery.
Most somatostatinomas (84%) have already metastasized at the time of presentation, but a number of patients survive 5 years after combined surgery and chemotherapy.
Among endocrine neoplasias, glucagonomas tend to be relatively large (5–10 cm) at diagnosis. In addition, up to 80% of such neoplasias show invasive or metastatic growth. Glucagonoma syndrome is recognized relatively late in most patients, and surgical treatment is possible in less than 20% of all patients. In this regard, such a pancreatic neuroendocrine tumor has the most serious prognosis.
Molecular subtypes of neuroendocrine pancreatic cancer
Modern molecular genetic methods make it possible to study tumors at the transcriptome level, and often as a result it turns out that one type of malignant neoplasm can be divided into several based on its molecular characteristics. This is what happened in a recent study by English and Italian scientists who sequenced transcriptomes of biopsy specimens of neuroendocrine pancreatic cancer [1]. As a result, 4 molecular subtypes were identified, differing from each other primarily in the activity of certain genes (Fig. 2).
Figure 2. Heat map showing the expression levels of immune genes (right) and a graph showing the percentage of immune genes expressed (left) in four subtypes of pancreatic neuroendocrine tumors. The most immune gene-rich subtype is metastasis-like primary tumor-1.
[1]
Immune cells (white blood cells) migrate to the neuroendocrine tumor, infiltrate it, and express immune-related genes.
The most “immune” of all molecular subtypes of pancreatic neuroendocrine tumors is metastasis-like primary tumor-1 ( MLP-1 ). The immunocytes found in it express 56% of immune genes (Fig. 2), which scientists divided into two groups (Fig. 3):
The first group is genes encoding activated T lymphocytes and cytokine proteins (interferons, interleukins, interferon stimulators).
The second group is the genes of monocytes/macrophages and dendritic cells, necessary for the processing of antigens.
Figure 3. Heat map depicting the expression levels of 19 identified immune genes in pancreatic neuroendocrine tumors. The top bar indicates the subtype. On the rainbow scale below the heat map, red (+1 point) means a high level of immune gene expression, blue (–1 point) means low, and white (0 points) means no gene expression. In the MLP-1 subtype, the expression of the genes LAG3, IFI16, SPP1, CCRL2, TREM2, ANXA1 is most pronounced.
[1]
Histological examination revealed that the MLP-1 tumor is less vascularized (poor in blood vessels) compared to other subtypes of pancreatic neuroendocrine tumors. Consequently, it is susceptible to hypoxia, which in turn stimulates programmed necrotic cell death - necroptosis. Unlike apoptosis, necroptosis is accompanied by a strong immune response: the dying cell releases damage-associated molecular patterns (damage-associated molecular patterns, DAMPs), which activate a cascade of immune reactions, especially the secretion of cytokines (types I and II interferons and interleukin 18 (IL-18)), mimicking the immune response to viral infection (viral mimicry). As a result, under hypoxic conditions, necroptosis of tumor cells increases, the number of DAMP molecules increases, and the immune response intensifies.
Necroptosis is programmed necrotic cell death, triggered through death receptors (for example, through the tumor necrosis factor receptor) and, unlike necrosis, has a clear regulatory system: it is regulated by protein kinases - enzymes that modify proteins. During necroptosis, as well as during apoptosis, the formation of reactive oxygen species in mitochondria occurs, but DNA fragmentation does not occur.
In search of an answer to the question of why this happens, researchers discovered that in the body of patients with neuroendocrine pancreatic cancer, dendritic cells (cells that introduce naïve T lymphocytes to antigens) intensively synthesize toll-like receptor 3 (TLR3), the ligand of which is a double-stranded RNA that appears during necroptosis. In addition, the number of dendritic cells increases (Fig. 4), therefore, the level of TLR3 expression increases, and this stimulates the synthesis of DAMP proteins.
Figure 4. Diagram showing the number of dendritic cells in four different molecular subtypes of pancreatic neuroendocrine tumors.
[1]
Based on the fact that necroptosis is necrotic cell death, for successful treatment it is necessary to exclude those conditions in which this will occur. A tumor is a living organ that feeds on oxygen. Lack of blood supply (ischemia) leads to hypoxia, which is the condition for the occurrence and intensification of necroptosis. Tumor cells (including neuroendocrine ones) have a unique ability to promote the appearance of new blood vessels (angiogenesis). However, such vessels are defective, they do not perform their function properly, as a result of which the tumor cells experience stress. Next, natural selection comes into play: vulnerable cells die, resistant cells migrate through the vessels to nearby lymph nodes, as well as to distant organs and tissues, forming secondary tumor foci - metastases (tumor clones). This process is called metastasis. It is one of the main strategies for tumor survival in the body. Stimulation of blood flow in the tumor promotes its intensive growth, which worsens the prognosis. However, this is the only way to avoid necroptosis, since DAMP molecules confuse the immune system, as a result of which it cannot destroy the tumor. A vicious circle, right?
As a result, in order to learn how to treat such tumors, we need to study in more detail the mechanisms of angiogenesis and analyze the level of cytokines and genes that are expressed by various cells of the tumor microenvironment in addition to immunocytes. It is also possible to identify new proteins that regulate necroptosis. All this in the long term will help defeat one of the most aggressive types of cancer.